




Your path to better sleep starts here

Understand my sleep health

Explore care options

Discover more about CPAP therapy
Looking for something else? We’re here to help — chat with Dawn, our 24/7 virtual assistant, or click here to learn more.

Dive into your sleep health
Learn more about the benefits of sleep, recommendations and our top tips.

CPAP mask categories to suit the way you sleep
Resmed CPAP mask categories are designed to reflect your needs and sleep habits. Our Versatile Fit, Freedom and Minimalist categories help make finding the right CPAP mask for you easier.
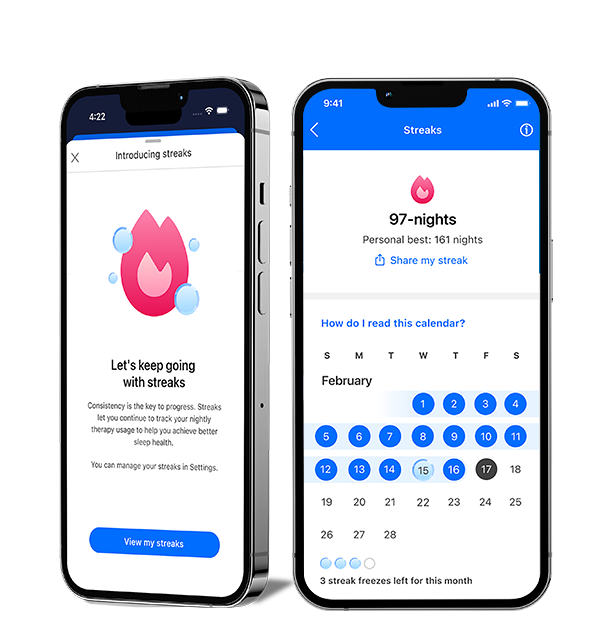
New to myAir: Streaks
See your progress and feel your momentum with the Streaks feature in myAir™. Easily track your consecutive nights on CPAP and celebrate your milestones. Start your streak tonight and set new records.

Residential Care Software
Our Residential Care Software team offers innovative, comprehensive software platforms that support healthcare providers in settings outside of the hospital.
Getting Started

A family that sleeps better, lives better

Tips for starting CPAP therapy
Stay in the know
Be among the first to hear all the latest news and developments.
* The mask contains magnets that may interfere with certain implants or medical devices. Please refer to the User Guide for complete labeling information, including magnet contraindications and warnings.